عبارت «ژنوم» به مواد ژنتیکی موجود زنده اشاره دارد. ژنوم یک ارگانیسم شامل کل DNA هستهای، از جمله نواحی کدکننده و غیر کدکننده، و همچنین DNA میتوکندری و کلروپلاست موجود در سلول است. مطالعه مواد ژنتیکی برای درک نقش مولکول DNA در سلول، «ژنومیک» نامیده میشود. در سال ۱۹۱۰، آلبرشت کسل، برنده جایزه نوبل، پنج باز نوکلئوتیدی یا «بازهای نوکلئوتیدی» را کشف کرد که اساس DNA و RNA، مواد ژنتیکی همه سلولهای زنده را تشکیل میدهند. این کشف به دنبال کشف الگوی جفتگیری چهار باز در DNA توسط اروین چارگاف در سال ۱۹۵۰ صورت گرفت که منجر به تعیین ساختار مارپیچ دوتایی DNA توسط جیمز واتسون و فرانسیس کریک، برندگان جایزه نوبل در سال ۱۹۵۳ شد.
تکنیکهای تعیین توالی DNA، که اولین بار در سال ۱۹۷۷ توسط فردریک سانگر (او نیز برنده جایزه نوبل است) توسعه یافت و کشف روش واکنش زنجیرهای پلیمراز (PCR) توسط کری مولیس (برنده دیگری از جایزه نوبل) در ۱۹۸۳، راه را برای تعیین توالی ژن و تحقیق برای یافتن ژنهای مرتبط با بیماریهای ژنتیکی مانند بیماری هانتینگتون، فیبروز کیستیک و هموفیلی در انسان هموار کرد. پروژه توالییابی کامل ژنوم انسان در سال ۱۹۹۰ آغاز شد، در حالی که اولین توالی کامل ژنوم یک ارگانیسم زنده آزاد، باکتری Haemophilus influenzae، در سال ۱۹۹۵ به دست آمد. از دهه ۱۹۹۰، با کمک تکنیکهای پیشرفته، امکانات و دانش در مورد DNA، توالی کامل ژنوم چندین ارگانیسم، از پروکاریوتها تا یوکاریوتها، منتشر شده است، از جمله توالی ژنوم انسان که در سال ۲۰۰۳ تکمیل شد.
کشف پلتفرمهای تعیین توالی نسل بعدی در سال ۲۰۰۸ هزینه تعیین توالی را به طور چشمگیری کاهش داد، در حالی که ترکیبی از تکنیکهای ویرایش ژنوم، تحقیقات ژنومی و ژنتیکی را متحول کرده است. توالی ژنوم موجود زنده که به صورت الگویی از چهار باز آدنین، تیمین، گوانین و سیتوزین (به اختصار “ATGC”) در DNA آن تشکیل شده است، اختصاصی بوده و میتواند تحتتأثیر عوامل درونی یا محیطی تغییر کند. توالی DNA یک ارگانیسم ممکن است طی همانندسازی DNA در تقسیم سلولی یا تحت تاثیر عوامل محیطی مانند اشعه ماوراء بنفش نور خورشید و قرار گرفتن در معرض مواد شیمیایی قادر به ایجاد جهش در DNA، تغییر کند. همچنین ممکن است برخی جهشهای ارثی در هر سلول وجود داشته باشد.
همه جهشهای DNA به بیماری مرتبط نیستند، بلکه در عوض اساس ژنتیکی ویژگیهای فنوتیپی متنوعی مانند گروه خونی و رنگ پوست، مو و چشم را تشکیل میدهند. این جهشهای DNA که باعث پروتئینهای غیرطبیعی نمیشوند و تنها مسئول تنوع فنوتیپی هستند، به عنوان نمونههایی از «چندریختی» شناخته میشوند. با این حال، برخی جهشهای DNA پروتئینهای غیرطبیعی تولید میکنند و باعث مشکلات جدی یا کشنده سلامتی میشوند. شناسایی خطاهای DNA عامل بیماری، دانشمندان را به فکر راههایی برای معکوس کردن این اشتباهات انداخت. اصلاح اختلالات مرتبط با ژنوم، دانشمندان را از اوایل دهه ۱۹۶۰ به دنبال استراتژیهای ویرایش ژنوم سوق داد.
ویرایش ژنوم به عنوان «تغییر عمدی و دقیق در یک ناحیه خاص هدفمند از توالی DNA در سلول» تعریف میشود .دانش در مورد مولکول DNA و در دسترس بودن تکنیکهای تعیین توالی DNA، امکان ویرایش مستقیم DNA در مکانهای دقیق و از پیش تعیین شده در میان بازهای آن را فراهم کرد. ویرایش ژنوم قبل از کشف نوکلئازهای قابل برنامهریزی DNA در دهه 1990 کار دشواری بود. با استفاده از تکنیکهای ویرایش ژنوم مبتنی بر نوکلئازهای DNA قابل برنامهریزی مجدد که در بازار موجود است و در این فصل توضیح داده میشود، تغییر دقیق در توالی نوکلئوتید (nt) از آن زمان به یک هدف واقعبینانه تبدیل شده است. به دلیل پیشرفتهای سریع و نوین در فناوری ویرایش ژنوم، اکنون وارد عصری میشویم که ویرایش ژنوم افق جدیدی را در تحقیقات پزشکی و کشاورزی به روی ما میگشاید.
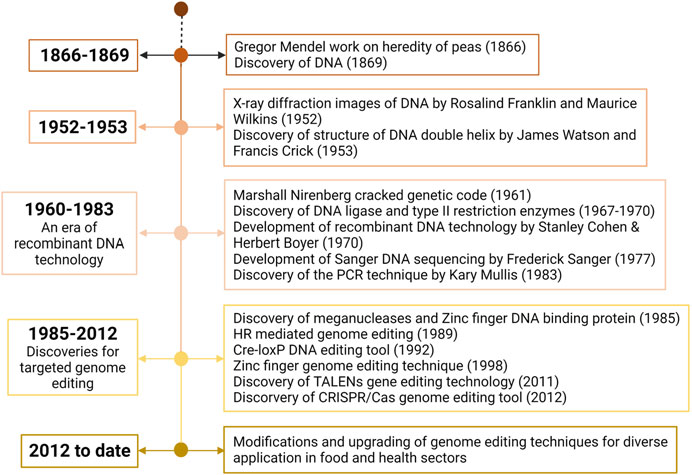
1.2 تاریخچه ویرایش ژنوم
ویرایش ژنوم یکی از با ارزشترین تکنیکهای موجود در تحقیقات بیولوژیکی است. برای درک کامل ارزش و جایگاه فعلی آن، درک تاریخچه این تکنیک بسیار مهم است. نقاط عطف متعددی در تاریخچه ویرایش ژنوم در شکل ۱.۱ نشان داده شده است. کشف و درک بعدی ساختار و عملکرد مولکول DNA قبل از ظهور فناوری DNA نوترکیب، راه را برای بسیاری از اکتشافات در ژنتیک و ویرایش ژنوم هموار کرد. کشف آنزیم لیگاز DNA و کشف آنزیمهای محدودکننده به ترتیب در سالهای ۱۹۶۷ و ۱۹۶۸، دو رویداد مهم در فناوری DNA نوترکیب و مهندسی ژنتیک بودند. این دو کشف با هم دنیای زیستشناسی را متحول کردند و فرصتی برای دستکاری DNA در آزمایشگاه (in vitro) ایجاد نمودند .متعاقباً، ظهور فناوری DNA نوترکیب، آغازگر تحقیقات دستکاری و ویرایش ژنوم بود .
شکل ۱.۱: نقاط عطف در تاریخ ویرایش ژنوم. این شکل رویدادهای کلیدی در تاریخ ژنومیکس را از کشف DNA تا ویرایش هدفمند ژنوم نشان میدهد. همچنین به پیشرفتهای مهمی اشاره میکند که امکان تبدیل پروتئینهای اتصالدهنده به DNA را به ابزارهای ویرایش دقیق ژن فراهم کرده است.
بنیان روشهای ویرایش ژن فرضیهای بود که شکستگیهای دو رشتهای هدفمند (DSB) در DNA که مسیرهای ترمیم داخلی سلولی DNA را تحریک میکند، میتواند برای ایجاد جهشهای هدفمند یا ویرایش دقیق در ژنوم مورد استفاده قرار گیرد .شکستگیهای دو رشتهای DNA به طور معمول برای سلول کشنده هستند. این شکستگیها میتوانند از طریق یکی از دو مسیر اصلی ترمیم شوند: ترمیم هدایتشده همسانی (HDR) یا اتصال انتهای غیرهمسان (NHEJ) در هر سلول همانطور که از نام آن پیداست، ترمیم هدایتشده همسانی (HDR) به بازترکیب توالی همسان بدون آسیب در یک کروماتید و در نتیجه ترمیم شکستگی به شیوه وابسته به الگو متکی است. از این مسیر میتوان برای گنجاندن DNA قالب اهداکننده همسانی که توسط محقق تأمین میشود، استفاده کرد (شکل ۱.۲ب). بنابراین، تغییرات دقیق و عمدی را میتوان در ژنوم ایجاد کرد که خاص DNA قالب خارجی (اگزوژن) است .
شکل ۱.۲: مکانیسمهای طبیعی ترمیم شکستگیهای دوطرفه DNA در سلولهای یوکاریوتی:
(الف) ترمیم اتصال انتهای غیرهمسان (NHEJ)
(ب) ترمیم هدایتشده توسط همسانی (HDR)
اتصال انتهای غیرهمسان (NHEJ) شکستگیهای دو رشتهای را با اتصال مستقیم انتهای شکسته به روشی غیر وابسته به الگو ترمیم میکند. این مسیر مستعد خطا است و اغلب منجر به طولهای متغیری از جهشهای درج و حذف (indels) در محل شکستگی میشود (شکل ۱.۲الف). یک شکستگی دو رشتهای با مکان خاص که در DNA ایجاد میشود، میتواند قاب خواندن باز (open reading frame) را توسط indels تغییر دهد، اگر این شکستگیها توسط NHEJ در سلول ترمیم شوند. بنابراین، ترمیم اشتباهی ایجاد شده از طریق NHEJ را میتوان برای ایجاد اختلال در ژن با مکان خاص با معرفی درج یا حذفهای کوچک پایه به کار برد با استفاده از مزیت ماشینآلات ترمیم داخلی سلول، ابزارهایی که شکستگیهای دو رشتهای ایجاد میکنند را میتوان برای تغییر دقیق ژنوم به کار برد.
مسئله چگونگی ایجاد شکستگیهای دو رشتهای (DSB) در محل هدف با کشف اندونوکلئازهای برش کمیاب طبیعی که به عنوان مگانوکلئازها شناخته میشوند در سال ۱۹۸۵ حل شد. مفهوم ویرایش ژنوم در اواخر دهه ۱۹۸۰ تثبیت شد. در سال ۱۹۸۹، کاپکی از بازترکیب همسان (HR) در سلولهای بنیادی جنینی موش برای هدف قرار دادن ژنهای خاص برای ایجاد سلولهای الحاقی (KI) و غیرفعالسازی (KO) استفاده کرد. اگرچه این فرآیند به دلیل فرکانس بسیار پایین HR در سلولهای پستانداران بسیار ناکارآمد بود، HR رویکردی انقلابی برای هدف قرار دادن ژنهای خاص در سلولهای یوکاریوتی ارائه کرد. از آنجایی که HR به تنهایی برای ادغام کارآمد ژن در سلولهای پستانداران کافی نیست و نیازمند انتخاب و غربالگری گسترده برای شناسایی یک سلول در میان یک میلیون سلولی است که ژن اصلاحشده را بیان میکند، معرفی شکستگیهای دو رشتهای به طور قابل توجهی باعث تسهیل بازترکیب میشود.
فناوری Cre-lox که بر اساس نوترکیبدهنده اختصاصی مکان DNA به نام Cre است، برای توسعه یک مدل موش تراریخته در اوایل دهه ۱۹۹۰ مورد استفاده قرار گرفت. این فناوری امکان غیرفعالسازی ژن هدف و فرصتی برای کنترل بیان ژن، به صورت مکانی و زمانی، مؤثرتر از HR به تنهایی را فراهم کرد. با این حال، به دلیل فاصله ژنتیکی بین مکانهای loxP، Cre-lox در کنترل برخی از ژنهای هدف کمتر مؤثر به نظر میرسید و از این رو این تکنیک نتوانست پذیرش گستردهای پیدا کند. با این حال، دانشمندان برای اولین بار از طریق این تکنیک توانایی دستکاری DNA و امکان ویرایش ژن با استفاده از اندونوکلئازها را به دست آوردند. از آن پس، از اندونوکلئازهای طبیعی برای ایجاد شکستگیهای دو رشتهای در DNA استفاده شد.
مگانوکلئازهای برش کمیاب که ۱۴ تا ۴۰ جفت باز (bp) را در DNA تشخیص میدهند، کارایی ویرایش ژنوم را افزایش دادند، اما مگانوکلئازهای طبیعی برای هر محل هدف در دسترس نبودند. بنابراین، دانشمندان شروع به بازمهندسی مگانوکلئازهای موجود برای DNA هدف کردند، اما کارایی هدفمند نوکلئازهای جهشیافته بسیار پایین بود. بعدها، کشف نوکلئازهای انگشت روی (ZFNs) در سال ۱۹۹۸، مگانوکلئازها را به عنوان یک تکنیک ویرایش ژن کنار زد. یک ZFN یک برشدهنده مصنوعی DNA است که از پروتئین اتصالدهنده DNA با انگشت روی به همراه اندونوکلئاز Fok1 تشکیل شده است. این تکنیک به محبوبیت گستردهای دست یافت، با این حال، یک پروتئین اتصالدهنده DNA دیگر به نام اثرگذار فعالکننده رونویسی (TALE) به راحتی قابل دستکاریتر از ZFNها شناخته شد.
از این رو، برای ساخت نوکلئاز هدفمند DNA، TALE با نوکلئاز Fok1 ترکیب شد و سیستم حاصلکننده «نوکلئازهای اثرگذار فعالکننده رونویسی» (TALENs) نامیده شد. در حالی که هر دو ZFN و TALEN مؤثر هستند، برای اصلاح ناحیه اتصال به DNA آنها برای هر هدف منفرد به منظور ایجاد شکستگیهای دو رشتهای (DSB) با مکان خاص برای ویرایش ژنوم، نیاز به تخصص در مهندسی پروتئین دارند. کشف «توالیهای تکرار کوتاه پالیندرومی فاصلهدار دستهبندیشده باکتریایی» (CRISPR)/Cas به عنوان ابزاری برای مهندسی ژنتیک در سال ۲۰۱۲ ویرایش ژنوم را متحول کرد. برخلاف ZFN و TALEN، CRISPR/Cas با استفاده از RNA راهنما به جای پروتئین، محلهای هدف را تشخیص میدهد.سیستم CRISPR/Cas شناختهشده امروزه از نوکلئاز Cas و RNA راهنما تشکیل شده است که با DNA هدف مکمل است. به دلیل سهولت طراحی و انعطافپذیری، این تکنیک به طور گسترده توسط جامعه علمی پذیرفته شده و در بسیاری از زمینههای علوم زیستی به کار گرفته میشود.
۱.۳تکنیکهای مختلف ویرایش ژنوم
تکنیکهای پیشرفته ویرایش ژنوم از نوکلئازهای طراحیشده و سیستمهای طبیعی ترمیم DNA موجود در سلولها برای ایجاد تغییرات دقیق در ژنوم استفاده میکنند. مکانیسمهای اثر برخی از قدرتمندترین تکنیکهای ویرایش ژنوم در اینجا مورد بحث قرار میگیرد.
1.3.1سیستم Cre-LoxP
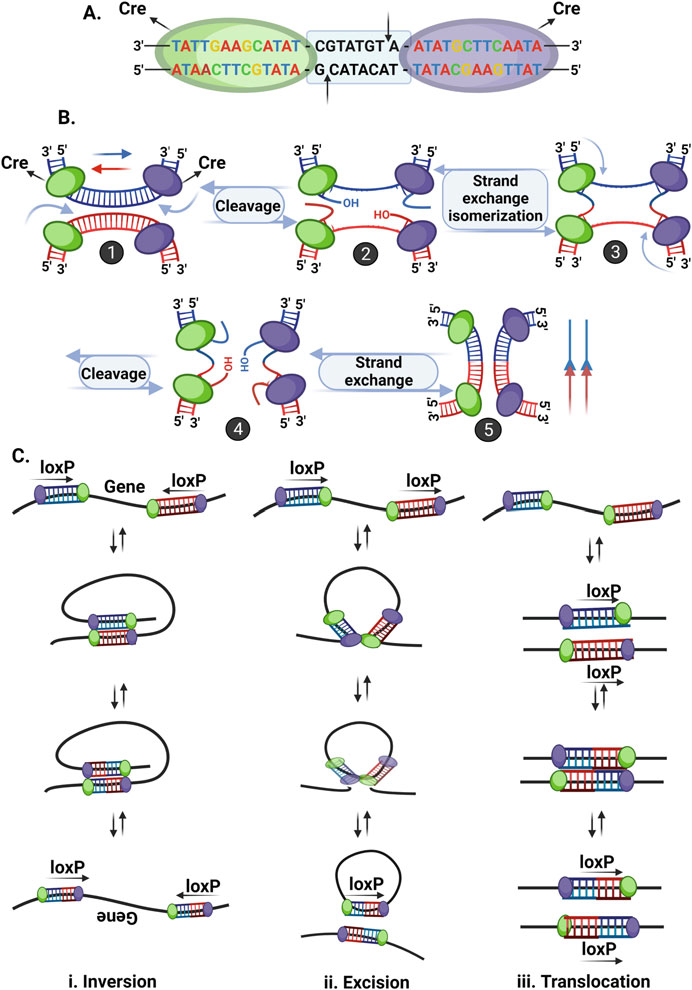
فناوری Cre-loxP در اوایل دهه ۱۹۹۰ به عنوان ابزاری کارآمد برای ویرایش ژن معرفی شد. این تکنیک بر اساس بازترکیب خاص مکان است که ویروسها به طور معمول برای ادغام DNA خود در ژنوم میزبان سازگار میشوند. این سیستم از دو مؤلفه عملکردی اقتباسشده از باکتریوفاژ P1 تشکیل شده است Cre یک نوترکیبدهنده و loxP محل تشخیص Cre. Cre یک نوترکیبدهنده ۳۸ کیلو دالتون (kDa) است که قطعات توالی خاص ۳۴ جفت باز DNA را که به عنوان محلهای loxP (مکان گذر، شناخته میشوند، تشخیص میدهد. محلهای loxP حاوی دو مجموعه ۱۳ جفت باز معکوس و پالیندروم هستند که با یک ناحیه فاصلهگذار ۸ جفت بازی از هم جدا شدهاند (شکل ۱.۳). توالی جفت باز در ناحیه فاصلهگذار متغیر است (به جز دو جفت باز در وسط)، که به توالی loxP جهت یا جهتگیری خاصی میدهد.
شکل ۱.۳: توالی ۳۴ جفت باز loxP. این شکل توالی ۳۴ جفت باز loxP را نشان میدهد که شامل دو بخش ۱۳ جفت باز تکراری پالیندروم با یک ناحیه فاصلهگذار است. "N" نشاندهنده بازهای متغیر در توالی ناحیه فاصلهگذار است.
اصول مونتاژ سیستم Cre-loxP و مکانیسم عمل آن در شکل ۱.۴ نشان داده شدهاست. دو نوترکیبدهنده Cre به هر ناحیه ۱۳ جفت بازی از توالی loxP متصل میشوند و یک دایمر را تشکیل میدهند (شکل ۱.۴الف). دو دایمر روی دو محل loxP با هم متصل میشوند تا یک تترامر را تشکیل دهند که محلهای loxP را با جهت مخالف به هم نزدیک میکند. پروتئین Cre، DNA دو رشتهای را در هر دو محل loxP میبرد و شکستگیهای دو رشتهای (DSB) ایجاد میکند که توسط آنزیم DNA لیگاز بسیار کارآمد ترمیم میشوند (شکل ۱.۴ب). در همین حال، یک رویداد گذر منجر به واژگونی، حذف و جابجایی توالی DNA میشود. مکان و جهتگیری محلهای loxP سه نوع بازآرایی مواد ژنتیکی را تعیین میکند (شکل ۱.۴ج):
شکل ۱.۴: مکانیسم عملکرد سیستم Cre-loxP
(الف) محلهای اتصال ۱۳ جفت بازی Cre (آنزیم نوترکیبدهنده) به توالی loxP که شامل یک ناحیه متغیر مرکزی است که شکستگی و تبادل رشته در آن رخ میدهد. شکستگی رشته بالایی بین T و A و شکستگی رشته پایینی بین جفت بازهای G و C است. (ب) مراحل مکانیسم واکنش (ج) واکنشهای تحت تأثیر جهتگیری توالی loxP: (i) اگر loxP در جهت مخالف باشد، Cre معکوسسازی قطعه DNA را تسهیل میکند. (ii) اگر loxP در جهت یکسان باشد، Cre باعث حذف (خارجسازی) قطعه DNA میشود. (iii) اگر loxP روی DNA های مختلف و در جهت یکسان باشد، Cre انتقال قطعه DNA را میانجیگری میکند.
وارونگی ( Inversion ): در صورتی که loxP روی رشتهی همانند DNA اما با جهت مخالف قرار داشته باشد، طی یک رویداد بازترکیبی، یک ژن وارونه میشود. این یک فرآیند برگشتپذیر است و ژنها میتوانند در هر زمانی به عقب و جلو برگردند.
حذف ( Excision ) : اگر محلهای loxP روی همان رشته DNA و در جهت یکسان قرار داشته باشند، یک توالی DNA قابل حذف است. در این حالت، یک ژن ممکن است به صورت برگشتناپذیری از محل اصلی خود توسط نوترکیبدهنده حذف شود.
جابجایی ( Translocation ) : در صورت وجود محلهای loxP روی رشتههای مختلف DNA، بازترکیبی منجر به جابجایی برگشتپذیر توالی DNA میشود.
به دلیل سهولت دستکاری، سیستم Cre-loxP به طور گستردهای برای ویرایش ژن در سلولهای پستانداران مورد استفاده قرار گرفته است. از این سیستم برای ایجاد مدلهای موش تراریخته در دهه ۱۹۹۰ استفاده شد. از آنجایی که Cre-loxP منحصراً در باکتریوفاژ وجود دارد و توالی loxP در سلولهای گیاهی یا حیوانی وجود ندارد، هنگام استفاده در سلولهای پستانداران، اتصال غیر اختصاصی نوترکیبدهنده Cre به حداقل میرسد. علاوه بر این، توالی بلند ۳۴ جفت بازی loxP احتمال اتصال غیر هدفمند پروتئین Cre را کاهش میدهد. این فناوری عمدتاً برای حذف ژن و ایجاد موشهای ناکارآمد (knockout) شرطی استفاده شده است.
سیستم Cre-loxP القاشدهنده با تتراسایکلین، CreERT، برای تنظیم بیان ژن مورد استفاده قرار گرفته و به عنوان ابزاری قدرتمند برای دستکاری شرطی ژن شناخته میشود. فناوری Cre-loxP به طور گسترده مورد استفاده قرار گرفته است: در عصبشناسی؛ در ایمونولوژی برای درک پاتوژنز ویروسی؛ در آنکولوژی (سرطانشناسی) و در مطالعات رفتاری و فیزیولوژیکی در موشها. در حالی که Cre-loxP این امکان را فراهم میکرد که بیان ژن را به راحتی بیشتری نسبت به HR کنترل کند، کارایی سیستم به دلیل فواصل ژنتیکی بزرگ بین محلهای loxP پایین بود. برخی وبسایتها اطلاعاتی در مورد سویههای موش Cre برای اهداف تحقیقاتی ارائه میدهند، اما هیچ پلتفرم مستقل، جهانی و واحدی برای جمعآوری یا به اشتراکگذاری آسان اطلاعات با محققان وجود ندارد.
علاوه بر این، توسعه سیستمهای تراریخته با درج ژن با فناوری Cre-loxP به زمان و تلاش قابل توجهی نیاز دارد. سیستمهای نوکلئاز مبتنی بر پروتئین جدیدتر، سادهتر و کارآمدتر به عنوان ابزارهای ژنتیکی برای ویرایش سریع و دقیق ژنوم با کاربردهای گستردهتر در علوم زیستی شناخته شدهاند.
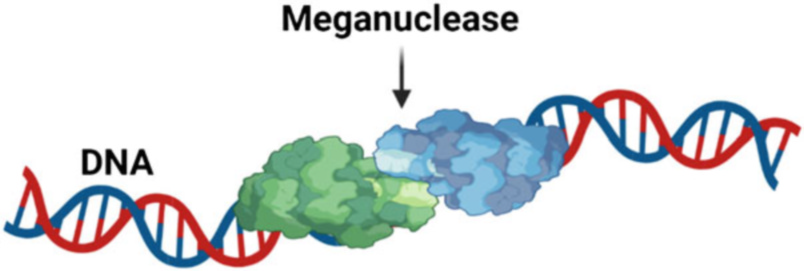
شکل ۱.۵: اتصال مگانوکلئاز به DNA هدف
1.3.2 مگانوکلئازها
مگانوکلئازها یا نوکلئازهای جهشیاب (HEs) اندونوکلئازهای طبیعی هستند که بر اساس توالی و نقوش ساختاری آنها به پنج خانواده تقسیم میشوند. این پنج خانواده عبارتند از: (۱) LADGLIDADG؛ (۲) HNH؛ (۳) GIY-YIG؛ (۴) His-Cys box؛ و (۵ PD-(D/E)XK. این خانوادهها از نظر مکانیسمهای کاتالیزوری، توزیع بیولوژیکی و شباهتها با نوکلئازهای غیر جهشیاب متفاوت هستند. مگانوکلئازها در حوزههای زیستی Archaea، باکتری Bacteria و یوکاریوتا Eukarya و ویروسهای مربوطه آنها یافت میشوند و به دلیل عملکرد ناشناختهشان در سلول، به عنوان «عناصر ژنتیکی خودخواه» شناخته میشوند. در بین این پنج خانواده، LADGLIDADG که به خانواده LHE نیز معروف است، به طور گسترده پراکنده و به خوبی شناخته شدهاست.
اعضای خانواده LHE به دلیل تماس زیاد DNA-پروتئین، ویژگی هدفمند بالا و انعطافپذیری، و توانایی اتصال به توالیهای طولانی DNA (۲۲ جفت باز یا بیشتر)، نسبت به اعضای سایر خانوادههای HE، گزینههای مناسبتری برای ویرایش ژنوم در نظر گرفته میشوند. اندونوکلئازهای خانواده LHE به صورت همودیمر و مونومر وجود دارند و توالیهای خاص DNA را از طریق برهمکنشهای پروتئین-DNA تشخیص میدهند. این اندونوکلئازها از طریق ورقه بتا پروتئین به بسیاری از بازهای DNA و ستون فقرات فسفات دی استر کناری در شیار اصلی متصل میشوند (شکل ۱.۵). بنابراین، تلاشهایی برای دستکاری برهمکنشهای پروتئین-DNA LADGLIDADG با تکنیکهای مهندسی پروتئین برای اهداف جدید صورت گرفت.
با وجود این، چندین مطالعه نشان دادهاند که پروتئینهای مرتبط میتوانند از زیرمجموعههای مختلفی از باقیماندهها برای تشخیص توالیهای DNA مشابه استفاده کنند. اتصال و شکستن DNA توسط مگانوکلئازها به هم وابسته است، به طوری که تقریبا ۵۰ اسید آمینه به طور مستقیم یا غیرمستقیم با DNA در تماس هستند که تغییر دادن ویژگی هدفمندی آنها به توالی مورد نظر DNA را با چالش قابل توجهی روبرو میکند. از این رو، بازمهندسی کردن ویژگی هدفمندی مگانوکلئازها دشوار است. با وجود این، چندین مورد شکافت اختصاصی مکان DNA توسط مگانوکلئازهای مهندسیشده، از جمله توالی ژنومی ذرت (گائو و همکاران ۲۰۱۰) و دو ژن انسانی XPC و RAG1 در منابع علمی گزارش شدهاست.
علاوه بر منشاء طبیعی، یکی از مزایای مگانوکلئازها برای ویرایش ژنوم، اندازه نسبتا کوچک (تقریباً ۴۰ کیلو دالتون) این اندونوکلئازها است که بستهبندی و انتقال آنها به سلولها را برای برخی کاربردها آسان میکند. یک مولکول هیبریدی به نام «مگاتال» که با ادغام یک اثرگذار TAL در حوزه DNA با مگانوکلئازها ایجاد میشود، برای اصلاح هدفمند ژن در سلولهای انسانی به کار رفته است. محفظه سازی درون شیشهای (IVC) با استفاده از چرخههای تکراری برای بازطراحی رابطهای گسترده پروتئین-DNA مگانوکلئازها به عنوان روشی کارآمد برای بازبرنامهریزی ویژگی هدف و شکافت مگانوکلئاز به کار گرفته شدهاست .
با این وجود، متیلدار شدن DNA و ساختار کروماتین که بر فعالیت نوکلئازی مگانوکلئازها تأثیر میگذارد، کاربردهای عملی این آنزیمها را در اپیژنتیک محدود میکند. اگرچه خط تولیدی مهندسی برای تغییر دادن ویژگیهای هدفمند مگانوکلئازها در مقیاس صنعتی توسعه یافته است، برای استفاده از این تکنیک در آزمایشگاه به دانش و تخصص ویژهای در مهندسی پروتئین نیاز است. علاوه بر این، احتمال بسیار زیاد اتصال غیرهدفمند، محدودیت دیگری برای این تکنیک است. پذیرش گستردهتر این فناوری همچنان به رویکردهای غیررسمی برای بازمهندسی این نوکلئازها برای اهداف مطلوب بستگی دارد. بنابراین، سایر تکنیکهای ویرایش ژنوم مانند ZFN و TALEN که از نظر بازبرنامهریزی برای توالیهای هدف جدید، در دسترس بودن منابع مهندسی، زمان و مقرون به صرفه بودن، انعطافپذیرتر هستند، نسبت به مگانوکلئازها ترجیح داده میشوند.
۱.۳.۳ نوکلئازهای انگشت روی (ZFNs)
ابزاری کارآمد و انعطافپذیر برای دستکاری DNA، نوکلئاز انگشت روی (ZFN) است که از دو جزء تشکیل شدهاست: یک حوزه اتصال به DNA قابل برنامهریزی و یک حوزه شکافت DNA. حوزه اتصال به DNA، پروتئینهای اتصال به DNA با انگشت روی روی (ZnF) هستند که توسط کلگ و همکاران در عامل رونویسی TFIIIa از تخم قورباغه (Xenopus) در سال ۱۹۸۵ کشف شد. انگشتهای روی روی فراوانترین گروه پروتئینها در انسان هستند و با بخش خاصی از DNA تعامل میکنند، بنابراین نقش اساسی در تنظیم رونویسی دارند. مطالعات نشان میدهند که هر ZnF حاوی نواحی تکرارشونده ۳۰ اسید آمینهای است که به شکل منحصربهفردی با دو ورقه بتا و یک مارپیچ آلفا تاخورده شدهاند.
این ساختار منحصربهفرد ββα توسط یک یون روی (Zn) که با دو جفت سیستئین و هیستیدین غیرقابل تغییر در ترکیبات مختلف اتصال برقرار میکند، حفظ میشود. ساختار کریستالی انگشتهای روی روی نشان میدهد که هر ZnF یک توالی ۳ تا ۴ جفت بازی خاص از DNA را تشخیص میدهد و با وارد کردن مارپیچ آلفا به شیار اصلی DNA به DNA متصل میشود. جالب اینجاست که شش اسید آمینه در مارپیچ آلفا در موقعیتهای ۱، +۱، +۲، +۳، +۵ و +۶ قابل تغییر هستند و میتوان از آنها برای ایجاد ویژگیهای نوین اختصاصی DNA در ZnF استفاده کرد، در حالی که سایر اسیدهای آمینهای محافظتشده به عنوان اسکلت ZnF عمل میکنند. بنابراین، ZnFها با تغییر اسیدهای آمینهای موجود در مارپیچ آلفا بر اساس توالی هدف DNA، یک چارچوب ایدهآل برای حوزه اتصال به DNA قابل برنامهریزی ارائه میدهند.
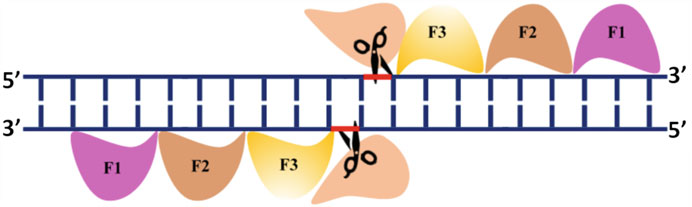
شکل ۱.۶: اتصال و برش DNA هدف توسط نوکلئاز انگشت روی (ZFN). این شکل نحوه اتصال و برش DNA هدف توسط نوکلئاز انگشت روی (ZFN) را نشان میدهد. یک جفت ZFN شامل سه تا شش پروتئین انگشت روی (ZFP) (حوزه اتصال به DNA) و آنزیم محدودکننده FokI (حوزه برش DNA) است که به هر دو رشته DNA هدف متصل شده و به دوطرفه شدن (دایمر شدن) FokI که برای فعالیت نوکلئازی FokI ضروری است، کمک میکند.
پس از شناسایی و اتصال به DNA هدف، شکافتن DNA دو رشتهای برای تحریک سیستمهای داخلی ترمیم DNA مورد نیاز است، که سپس ممکن است در طول ترمیم شکستگیهای دو رشتهای (DSB) به تغییراتی در آن مکان خاص منجر شود. بنابراین، برای ایجاد شکستگیهای دو رشتهای جهت ویرایش دقیق ژنوم، اتصال یک حوزه شکافت DNA به یک حوزه اتصال به DNA ضروری است. مشاهدات نشان دادهاند که آنزیمهای محدود کننده نوع دوم مانند BamHI و EcoRI به دلیل همپوشانی بین عملکرد شناسایی و محدود کردن DNA، به عنوان ابزارهای ویرایش ژنوم مناسب نیستند. تغییر اسیدهای آمینهای اتصال به DNA برای تغییر ویژگی هدفمندی آنزیمهای محدود کننده نوع دوم، باعث تغییر محل کاتالیزوری این آنزیمها میشود و آنزیمهای غیرفعال از نظر کاتالیزوری تولید میکند.
با این حال، آنزیم محدود کننده نوع IIS FokI نه تنها پنج باز DNA غیربسامد 5′-GGATG-3′ را در دو رشته 5′-CATCC-3′ تشخیص میدهد، بلکه حاوی دو حوزه جداگانه است: حوزه اتصال به DNA انتهای N ترمینال ۴۱ کیلو دالتون و حوزه شکافت DNA انتهای C ترمینال ۲۵ کیلو دالتون. به طور معمول، اندونوکلئاز FokI، ۹/۱۳ نوکلئوتید پایینتر از توالی شناسایی را میشکافد، اما افزودن اتصالدهندههای ۴ اسید آمینهای بین ۲ حوزه، محل برش را ۱ جفت باز به پایینتر از محل برش اصلی منتقل میکند، یعنی به جای ۹/۱۳ جفت باز در آنزیم طبیعی، به ۱۰/۱۴ جفت باز منتقل میشود. بنابراین، یک نوکلئاز کایمریک با پیوند زدن حوزه شکافت FokI با پروتئینهای انگشت روی اتصال به DNA برای ایجاد یک نوکلئاز جدید با ویژگی اختصاصی به توالی ساخته شد تا DNA هدف را برش دهد .
به طور معمول، سه تا شش انگشت روی روی باهم به صورت تکرار پشت سر هم متصل میشوند تا یک پروتئین انگشت روی (ZFP) را تولید کنند که به یک محل هدف DNA به طول ۹ تا ۱۸ جفت باز متصل میشود. ادغام ZFPهای سفارشیسازیشده با نوکلئاز کایمریک FokI، نوکلئازی طراح (designer nuclease) ایجاد میکند که DNA را در محلهای هدف خاص برش میدهد. از آنجایی که FokI برای فعالیت نوکلئازی خود به دایمر شدن نیاز دارد، شکافت DNA دو رشتهای نیازمند اتصال دو نسخه از ZFPهای اختصاصی به توالی در رشتههای مخالف DNA در جهت دم به دم معکوس است که باعث دایمر شدن FokI برای ایجاد شکستگیهای دو رشتهای (DSB) در DNA میشود (شکل ۱.۶).
هنگامی که یک جفت ZFP به DNA هدف خاص در رشته مستقیم و معکوس متصل میشود، FokI دایمر میشود تا شکستگیهای دو رشتهای DNA را ایجاد کند که مکانیسم ترمیم DNA درونزا را تحریک میکند. بنابراین، جهشهای کوچک در بازها، حذفها یا خاموش کردن ژن میتواند از طریق مکانیسمهای مختلف ترمیم DNA و با کارایی کم اتفاق بیفتد. هنگامی که یک جفت ZFP به DNA هدف خاص در رشته مستقیم و معکوس متصل میشود، FokI دایمر میشود تا شکستگیهای دو رشتهای DNA را ایجاد کند که مکانیسم ترمیم DNA درونزا را تحریک میکند. بنابراین، جهشهای کوچک در بازها، حذفها یا خاموش کردن ژن میتواند از طریق اتصال انتهایی غیر همسان (NHEJ) یا در حضور قالب دهنده، ژنی با واسطه ترمیم نوترکیبی هومولوژی هدفی (HDR) وارد شود.
ادغام ZFPها با حوزه نوکلئازی FokI نه بر توانایی اتصال ZFPها به محل هدف تأثیر میگذارد و نه فعالیت محدودکنندگی حوزه نوکلئازی FokI را از بین میبرد. بنابراین، این تکنیک به طور گستردهای برای ویرایش ژنوم در حیوانات و گیاهان پذیرفته شدهاست. همچنین با ادغام ZFPها با حوزههای فعالکننده یا سرکوبکننده، فعالکنندههای انگشت روی (ZFAs) و سرکوبکنندههای انگشت روی (ZFRs) ایجاد شدهاند که برای فعال یا سرکوب ژن خاص به یک محل شناسایی تنها در DNA هدف متصل میشوند.
طراحی و انتخاب ZFPهای با ویژگی هدفمندی بالا برای توالی DNA هدف در ژنوم پیچیده، کاری پرزحمت و زمانبر است. شناسایی هدف توسط ZnFها تحت تأثیر شدید نوtifهای انگشت روی مجاور قرار دارد. وجود باقیمانده آسپارتات در موقعیت +۲ در ساختار مارپیچ آلفای یک نوتیف ZnF، شناسایی ZnF را از ۳ به ۴ جفت باز تغییر میدهد که بر ویژگی اختصاصی ZnFهمسایه در آرایه تأثیر میگذارد. بنابراین، طراحی و انتخاب آرایههای ZnF زمانبر و چالشبرانگیز است و منجر به پیشبینیناپذیری در ویژگی هدفمندی در آرایه نهایی میشود. علاوه بر این، سمیت ZFNها به دلیل شکافت خارجهدف، مسئله جدی دیگری است که کاربرد این فناوری را در درمانهای انسانی محدود کردهاست.
تلاشهایی برای بهبود این فناوری صورت گرفتهاست. مشاهده شدهاست که اتصال خارجهدف ZFNها وابسته به غلظت است؛ این را میتوان با استفاده از وکتور عباس با بیان کنترلشده کاهش داد. علاوه بر این، برای بهبود ویژگی هدفمندی، تعداد ZnFها در ZFNها میتواند افزایش یابد. همچنین، میتوان ZFPهای با ویژگی هدفمندی بالا را برای دستیابی به میل ترکیبی بالا با DNA هدف و کاهش سمیت ZFNها در شرایط آزمایشگاهی (in vivo) بررسی کرد. با این وجود، پس از آشکار شدن محدودیتهای این فناوری، محققان شروع به جستجوی استراتژیهای جایگزین کردند که منجر به کشف فناوریهای ویرایش ژنوم قدرتمند به نام TALENها و CRISPR/Cas9 شد که در ادامه به آنها پرداخته میشود.
۱.۳.۴ نوکلئازهای اثرگر شبه فعالساز رونویسی (TALENs)
TALEN یک ابزار ویرایش ژنوم قابل برنامهریزی است که بر اساس ماژول جدید اتصال به DNA به نام پروتئین اثرگر شبه فعالساز رونویسی (TALE) که با حوزه کاتالیتیکی اندونوکلئاز FokI ادغام شدهاست، بنا شدهاست. پروتئین TALE در باکتریهای بیماریزای گیاهی از جنس Xanthomonas یافت شد. عملکرد ذاتی پروتئینهای TALE اتصال به DNA ژنومی میزبان و تغییر رونویسی ژنهای آنها برای تسهیل استعمار باکتریهای بیماریزا در میزبان است. پروتئین TALE از ۳۳ تا ۳۵ توالی تکرارشونده با ۳۳ تا ۳۵ اسید آمینه با نگهداری بالا تشکیل شدهاست که در هر تکرار دو اسید آمینه بسیار متغیر در موقعیتهای ۱۲ و ۱۳ وجود دارد.
شکل ۱.۷: اتصال و مکانیسم اثر نوکلئاز اثرگذار شبیه فعالگر رونویسی (TALEN). شکل نشاندهنده اتصال و مکانیسم اثر نوکلئاز اثرگذار شبیه فعالگر رونویسی (TALEN) است. یک TALEN از مونومرهای چپ و راست پروتئینهای TALE (حوزه اتصال به DNA) و آنزیم محدودکننده FokI (حوزه برش DNA) تشکیل شده است که در صورت تشکیل دایمر، DNA را برش میدهد. هر TALE یک جفت باز واحد از توالی هدف DNA را تشخیص میدهد.
این دو اسید آمینه متغیر، که باقیماندههای متغیر تکراری دوگانه (RVDs) نامیده میشوند، بازهای خاصی از DNA را تشخیص میدهند، برای مثال RVDهایی مانند آسپارژین و ایزولوسین (NI)، آسپارژین و گلیسین (NG)، دو آسپارژین (NN) و هیستیدین و آسپارتیک اسید (HD) به ترتیب به نوکلئوتیدهای A، T، G و C متصل میشوند. برخلاف انگشتهای روی روی که با سه نوکلئوتید متصل میشوند، یک تکرار TALE تنها یک نوکلئوتید را تشخیص میدهد (شکل ۱.۷). ساختار کریستالی TALE متصل به هدف DNA آن نشان میدهد که باقیماندههای دوگانه متغیر در هر تکرار، یک موقعیت را در شیار اصلی DNA اشغال میکنند و اسید آمینه در موقعیت ۱۳ با DNA تماس اختصاصی با نوکلئوتید برقرار میکند، در حالی که اسید آمینه در موقعیت ۱۲ با ایجاد تماس با اسید آمینه در موقعیت ۸ در دامنه، ثبات ساختاری ایجاد میکند.
تغییر در RVDها ویژگی هر تکرار را نسبت به نوکلئوتید هدف خود تغییر میدهد، بنابراین آرایهای از TALEها میتوانند به توالی طولانیتر DNA به طول ۳۰ تا ۴۰ جفت باز متصل شوند. ویژگی اتصال به DNA هر تکرار TALE مستقل بوده و بر ویژگی اتصال TALEهای مجاور تأثیر نمیگذارد و مطابقت یک به یک بین تکرارهای TALE و توالی هدف DNA را فراهم میکند. تعداد تکرارها در یک آرایه به طول توالی هدف بستگی دارد. مکانیسم سادهی تشخیص DNA توسط TALEها آنها را به گزینهای ایدهآل برای ساختن یک نوکلئاز سفارشی تبدیل میکند. ادغام ماژول قابل تنظیم اتصال به DNA از TALE با نوکلئازهای FokI نشاندهندهی یک تکنیک ویرایش ژنوم با اهمیت گسترده در علوم زیستی بود و در سال ۲۰۱۱ توسط «Nature Methods» به عنوان «روش سال» توصیف شد.
مکانیزم کلی عملکرد TALENها شبیه به ZFNها است. حوزه TALE با ویژگی هدفمندی، هدف DNA را در ژنوم تشخیص میدهد و حوزه غیر اختصاصی نوکلئاز FokI پس از دایمر شدن، DNA را میشکافد و DSBهای هدفمند ایجاد میکند (شکل ۱.۷) که برای ویرایش ژنوم ضروری است. از آنجایی که شناسایی هدف توسط TALENها به یک نوکلئوتید محدود میشود، TALENها نسبت به ZFNها از نظر هدف ویژهتری برخوردار هستند، در حالی که در ZFNها، ویژگی تحت تأثیر قابل توجهی یک موتیف ZF مجاور قرار دارد.
۱.۳.۵ سیستم CRISPR/Cas
ارگانیسمها در قلمروهای زیستی باکتری، آرکیباستان و یوکاریوتا دائماً در معرض مواد ژنتیکی خارجی از منابع مختلف از جمله باکتریوفاژها، ترانسپوزونها و پلازمیدها قرار دارند. در نتیجه، آنها به طور طبیعی برای محافظت از خود در برابر مهاجمان، سازوکارهای دفاعی را تطبیق میدهند. یکی از چنین نوع سیستمهای ایمنی طبیعی توسط باکتریها و آرکیباستان تطبیق داده شده، سیستم CRISPR/Cas است. تحقیقات نشان میدهد که CRISPR/Cas به ترتیب تقریباً در ۴۰٪ باکتریها و ۹۰٪ سیستمهای ایمنی آرکیباستانها وجود دارد.
این سیستم دفاعی طبیعی بین پروکاریوتها به طور منحصر با مولکولهای RNAکوتاه (crRNA) و پروتئین خاص Cas کار میکند. crRNA فعالکننده ترانس، DNA خارجی را تشخیص میدهد و پروتئین Cas را برای اتصال به DNA مهاجم هدایت میکند و سپس ممکن است این DNA توسط فعالیت نوکلئازی Cas شکافته شود. هم crRNA و هم اعضای مختلف خانواده پروتئین Cas یک کمپلکس اثرگذار ایجاد میکنند که نه تنها DNA خارجی را شناسایی میکند بلکه شکستگیهایی را در محل خاصی از DNA ایجاد میکند تا آن را تخریب کند.
شناسایی DNA خارجی از طریق CRISPR/Cas به وجود توالی خاصی از آن DNA در crRNA نیاز دارد که این توالی به عنوان «فاصلهزن» (spacer) نامیده میشود. فاصلهزن معمولاً از برخورد قبلی میزبان با آن DNA خارجی به دست میآید. با یک عفونت مجدد، crRNA سیستم CRISPR توالی مطابق مهاجم را که به عنوان «پیش-فاصلهزن» (protospacer) شناخته میشود، از طریق جفت شدن بازهای متمم شناسایی میکند و به آن متصل میشود و اجازه میدهد تا پروتئین Cas از طریق فعالیت نوکلئازی خود، DNA دو رشتهای را متصل کند و در نهایت مهاجم را غیرفعال کند.
درک مکانیسم عمل CRISPR/Cas در میکروارگانیسمهای مختلف به دانشمندان اجازه داده است تا از آن به عنوان تکنیکی برای دستکاری DNA استفاده کنند. کلاسهای متمایز سیستم CRISPR/Cas بر اساس تغییرات ساختاری و سبک سازمانی ژنهای cas شناسایی شدهاند و این پیشرفتها به تبدیل این سیستم به یک ابزار ویرایش ژنوم ضروری کمک کردهاند.
۱.۳.۵.۱ مکانیسم عمل سیستم CRISPR/Cas
به طور کلی، مکانیسم عمل سیستم CRISPR/Cas به سه مرحله تقسیم میشود.
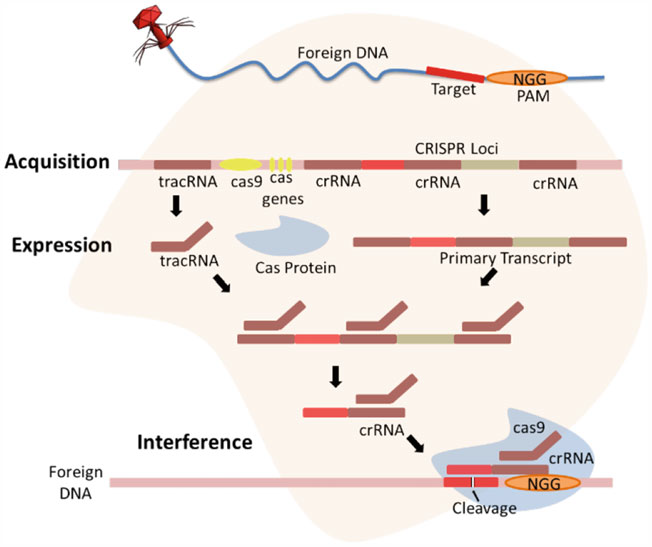
مرحله اول، مرحلهی ایمنیسازی است که به عنوان مرحلهی «سازگاری» یا «کسب فاصلهزن» نیز شناخته میشود. در طول این مرحله، یک سلول باکتریایی با گرفتن توالی کوتاه DNA از ژنوم مهاجم و ادغام آن به عنوان فاصلهزن در جایگاه کریسپر سلول، خود را با DNA مهاجم تطبیق میدهد. هر فاصلهزن جدید در ابتدای آرایهی CRISPR، در کنار یک توالی پیشرو غنی از AT قرار میگیرد. به طور کلی، جایگاه ژنومی CRISPR/Cas از مجموعهای از ژنهای cas، یک توالی پیشرو و عناصر تکرارشونده کوتاه (تکرارها) که توسط فاصلهزنهای منحصربهفرد از هم جدا شدهاند، تشکیل شدهاست (شکل ۱.۸). فاصلهزن به عنوان یک رکورد ژنتیکی از عفونتهای قبلی عمل میکند که حافظهی ایمنی در میزبان برای شناسایی مهاجم در آینده ایجاد میکند. شواهد تجربی نشان میدهد که سازگاری و ادغام فاصلهزن نیازمند پروتئینهای Cas1 و Cas2 است. مشاهده شدهاست که حذف پروتئینهای Cas1 و Cas2 از سیستم، فرآیند سازگاری را بدون تأثیر بر پاسخ ایمنی کریپسر متوقف میکند، که نشاندهندهی نقش پروتئینهای Cas1 و Cas2 در مکانیسم «کسب فاصلهزن» است.
شکل ۱.۸: سه مرحله از مکانیزم اثر سیستم ایمنی طبیعی CRISPR-Cas. این شکل سه مرحله از عملکرد سیستم ایمنی طبیعی CRISPR-Cas را نشان میدهد: ۱. اکتساب (Acquisition):در این مرحله، باکتری قطعهای از DNA مهاجم (ویروس یا پلاسمید) را به عنوان یک (spacer) انتخاب کرده و آن را درون جایگاه CRISPR (CRISPR locus) در ژنوم خود جایگذاری میکند. ۲. بیان (Expression): در مرحله بیان، ناحیه CRISPR با کمک RNA پلیمراز رونویسی شده و یک RNA پیشساز بلند (pre-crRNA) تولید میشود. سپس این RNA پیشساز پردازش شده و به RNA راهنما (guide RNA) که شامل توالی spacer است، تبدیل میشود. ۳. تداخل (Interference): در صورت بازعفونت، مرحله تداخل رخ میدهد. در این مرحله، RNA راهنما با استفاده از توالی خاص PAM (توالی مجاور پروتواسپاسر) که در بالا یا پایین پروتواسپاسر در DNA هدف (DNA پاتوژن) قرار دارد، DNA هدف را شناسایی میکند. پس از شناسایی، پروتئین Cas نوکلئاز (Cas nuclease) موجود در کمپلکس CRISPR-Cas، DNA هدف را برش میدهد و از عفونت جلوگیری میکند.
مرحله دوم، مرحلهی ایمنی به نام «بیان و بلوغ» شناخته میشود. «بیان» به رونویسی آرایهی کریپسر به یک پیشمادهی RNA بلند (pre-crRNA) اشاره دارد، در حالی که «بلوغ» پردازش بعدی pre-crRNA برای ساخت یک crRNA راهنمای بالغ حاوی توالیهای فاصلهزن است. پردازش pre-crRNA در سیستم کریپسر نوع II نیازمند اتصال pre-crRNAبا یک tracer RNA (RNA ردیاب) کوتاه غیر کدکننده ترانسفعالشده از طریق توالیهای ضدتکرار tracer RNA است که یک دو رشتهای RNA ایجاد میکند. این دو رشتهای RNA توسط پروتئین Cas9 تثبیت میشود و میتواند توسط RNA III درونزا برای تولید یک crRNA واسط شناسایی شود که تحت فرآیندی با جزئیات ناشناخته بیشتر بالغ میشود تا یک crRNA راهنمای بالغ کوچک (gRNA) تولید شود. crRNAبالغ در انتهای ۳’ خود دارای بخش کوچکی از عنصر تکرارشونده (تکرار) و در انتهای ۵’ خود دارای یک توالی فاصلهزن کامل است که به tracer RNA و Cas9متصل میماند تا یک کمپلکس Cas-crRNA فعال را تشکیل دهد (شکل ۱.۸).
شکل ۱.۹: نمونهای از چگونگی کلی مونتاژ و شناسایی هدف در ابزار ویرایش ژنوم CRISPR/Cas. این شکل یک RNA راهنمای تک (sgRNA) را نشان میدهد که از دو بخش تشکیل شده است: توالی crRNA: این توالی اختصاصی برای هدف DNA است و به شناسایی محل برش در DNA هدف کمک میکند. توالی ردیاب RNA: این توالی با پروتئین Cas9 که فعالیت نوکلئاز اندونوکلئازی (برشدهندهی دوطرفه DNA) دارد، برهمکنش میکند. مجموع این اجزا، کمپلکس CRISPR/Cas را تشکیل میدهند که باعث شکستگی دوطرفه و هدفمند DNA میشود و این شکستگی برای ویرایش ژنوم ضروری است.
مرحله سوم، ایمنی CRISPR/Cas به عنوان «تداخل» شناخته میشود. کمپلکس Cas-crRNAسلول را برای اهداف DNA خارجی اسکن میکند و با شناسایی توالی مکمل فاصلهزن در ژنوم مهاجم، یعنی «پیش-فاصلهزن» (protospacer)، با عوامل بیماریزای مهاجم تداخل میکند. هنگامی که پیش-فاصلهزن در DNA هدف توسط فاصلهزن از طریق جفت شدن بازهای مکمل شناسایی میشود، کمپلکس Cas-crRNA پیش-فاصلهزن را با فعالیت نوکلئازی میشکافد که منجر به تخریب DNA خارجی میشود. در اینجا مهم است که توجه داشته باشید که شناسایی هدف و شکافت DNA هدف توسط یک کمپلکس Cas-crRNA فعال به وجود یک موتیف کوتاه مجاور پیشفاصلهزن (PAM)در کنار توالی پیش-فاصلهزن بستگی دارد (شکل ۱.۹). PAM یک توالی محافظتشده ۲ تا ۵ نوکلئوتیدی است که توسط پروتئین Cas شناسایی میشود و نقش مهمی در شناسایی هدف و تمایز بین DNA خودی و غیرخودی برای جلوگیری از خود ایمنی ایفا میکند. پیشرفتها در درک مکانیسم CRISPR/Cas (شکل ۱.۸ و ۱.۹) دانشمندان را قادر ساخته است تا این سیستم را به یک ابزار ویرایش ژنوم بسیار کارآمد و دقیق تبدیل کنند.
۱.۴ ظهور سیستم CRISPR/Cas
کشف ابزارهای مهندسی ژنوم مانند مگانوکلئازها و سپس ZFNها و TALENها، دستکاری هدفمند DNA را تسهیل کرده و به درک نحوه تنظیم ژن و ارتباط بین اختلالات ژنتیکی و بیماریهای خاص کمک کرده است. هر دو نوکلئاز طراحیشده، ZFNها و TALENها، به ترتیب بر اساس شناسایی DNA هدف قابل اصلاح اتصال به DNA مانند انگشت روی روی و حوزههای TALE عمل میکنند. به دلیل مشکلات بازطراحی پروتئینهای ماژولار برای هر DNA هدف، محققان به دنبال رویکردهای جایگزینی بودند که سهولت و ماژولار بودن بازمهندسی ابزار ویرایش ژنوم و همچنین ویژگی هدفمندی را ارائه دهند. کشف اخیر سیستم کریپسر به دلیل مزایای متعدد آن (مذکور در بخش ۱.۵) نسبت به ZFNها و TALENها، این حوزه را متحول کرد.
سیستم محبوب نوع II CRISPR/Cas9 که از استرپتوکوکوس پیوژنز به دست آمده است، به دلیل سهولت طراحی و دستکاری برای ویرایش ژنوم هدف، به طور گسترده مورد بررسی و پذیرش قرار گرفتهاست. CRISPR/Cas به عنوان یک «سیستم ذخیرهسازی حافظه» برای محدود کردن عفونت مجدد توسط عوامل بیماریزا در پروکاریوتها عمل میکند. یافتههای کلیدی در سالهای اخیر، سیستم ایمنی کریپسر را به یک ابزار قدرتمند قابل برنامهریزی برای ویرایش ژن تبدیل کرده است. وجود تکرارهای CRISPR برای اولین بار توسط گروه آتسوئو ناکاتا در سال ۱۹۸۷ در DNA اشریشیاکلی مشاهده شد، مدتها قبل از اینکه نام «CRISPR» ابداع شود. پس از آن، شناسایی ماهیت و منشأتوالی فاصلهزن از طریق تجزیه و تحلیل محاسباتی توالیهای کامل ژنوم فاژها و سایر موجودات، محققان را متوجه این نکته کرد که توالیهای فاصلهزن متعلق به باکتریوفاژها و سایر عناصر ژنتیکی متحرک هستند. علاوه بر این، وجود چندین ژن مرتبط با پروتئین Cas محافظتشده و مرتبط با CRISPR، علاقه محققان را برای بررسی عملکرد بالقوه و مکانیسم عمل کل آبشار کریپسر افزایش داد.
اهمیت عملکردی سیستم کریپسر توسط Horvath روشن شد، که شواهد تجربی همبستگی بین فعالیت کریپسر و پاسخ ایمنی تطبیقی در پروکاریوتها را ارائه داد. آنها وجود یک توالی فاصلهزن جدید در محل CRISPR استرپتوکوکوس ترموفیلوس بعد از مواجهه با ویروس را مشاهده کردند. جالب اینجاست که توالی فاصلهزن جدید بهدستآمده توسط باکتری متعلق به توالی ژنومی باکتریوفاژ بود که منجر به ایجاد سویه استرپتوکوکوس ترموفیلوس مقاوم به باکتریوفاژ شد. کسب توالی فاصلهزن پیشنیاز اختصاصیت هدفگیری مستقیم نوکلئازهای Cas بود که سیستم دفاعی منحصربهفردی را در برابر باکتریوفاژ ایجاد میکند. پس از این کشف کلیدی، محققان بیشتر متوجه شدند که برای هدایت نوکلئازهای Cas به هدفشان، به RNAهای کوتاه کریپسر رونویسیشده از توالیهای فاصلهزن نیاز است.
یافته مهم دیگری، شناسایی PAMها بهعنوان یک نیاز ضروری برای عملکرد سیستم کریپسر بود. مشاهده مهمی که توسط ساپراناسکاس و همکارانش انجام شد، توانایی سیستم کریپسر برای انتقال از یک باکتری به گونههای باکتریایی دیگر بود. این امر محققان را برای مشخص کردن بیشتر اجزای منفرد سیستم CRISPR به صورت بیوشیمیایی و درک دقیقتر مکانیسم مولکولی آن تحریک کرد. یک یافته مهم این بود که در میان چندین پروتئین Cas، تنها Cas9 از S. thermophilus دارای فعالیت کاتالیزوری DNA بود؛ علاوه بر این، برای عملکرد خود به دو RNA کوتاه نیاز داشت.
این یافتهها، به همراه مشاهدهی قابلیت برنامهریزی مجدد Cas9 مطابق با توالی هدف DNA ، آغاز CRISPR/Cas بهعنوان یک ابزار ویرایش ژنوم قابل برنامهریزی مجدد را رقم زد. ساخت یک sgRNA کایمریک با ادغام دو RNA راهنمای کوتاه (tracer RNA و crRNA) برای هدایت Cas9 به هدف، طراحی فناوری CRISPR/Cas را برای اهداف خاص ساده کرد. این امر با سازگاری پیشگامانۀ این ابزار برای ویرایش ژنوم در سلولهای یوکاریوتی با کارایی، ویژگی و انعطافپذیری بالا دنبال شد. از زمان آغاز به کار CRISPR/Cas به عنوان یک ابزار ویرایش ژنوم، محققان از زمینههای مختلف از این فناوری برای بازسازی دقیق ژنومهای پروکاریوتی و یوکاریوتی استفاده کردهاند و کاربردهای بالقوه آن را در تمام زمینههای زیستفناوری، از جمله کاربردهای ضد میکروبی، درمانی و تشخیصی نشان دادهاند.
۱.۵ مقایسه سیستم کریسپر کس با سایر ابزارهای ویرایش ژنوم
ابزارهای ویرایش ژنوم با نوکلئازهای قابل برنامهریزی به دلیل کاربردهای بالقوه آنها در زمینههای ژنتیک، زیستفناوری و درمان، توجه زیادی را به خود جلب کردهاند. تلاش و تحقیقات مداوم به محققان اجازه داده است تا نامزدهای جدید ویرایش ژنوم را کشف کرده و خواص ابزارهای موجود را بهبود بخشند. توجه اصلی در حال حاضر بر توسعه تکنیکهای ویرایش ژنوم ساده، مقرون به صرفه و با هدفمندی بالا با کاربردهای گسترده در علوم پایه و زیستپزشکی متمرکز است.
هر چهار تکنیک ویرایش ژنوم که تاکنون معرفی شدهاند (مگانوکلئازها، ZFNها، TALENها و CRISPR/Cas9) برای اصلاح ژنوم هدفمند در ارگانیسمهای مدل، گیاهان و ردههای سلولی انسانی به طور مؤثری مورد استفاده قرار گرفتهاند. با این حال، پاسخ به این سؤال که کدام فناوری قابل برنامهریزی مجدد برای ویرایش ژنوم برای همه موارد مناسبتر است، دشوار است. کاربرد هر ابزار ویرایش ژنوم به کارایی، ویژگی هدفمندی، سادگی طراحی، هزینه و قابلیت اطمینان آن بستگی دارد. مقایسهای از نوکلئازهای قابل برنامهریزی در جدول ۱.۱ نشان داده شدهاست (به متن اصلی برای جدول ۱.۱ مراجعه کنید). در حالی که ZFNها و TALENها مؤثر هستند، طراحی آنها نیازمند تخصص در مهندسی پروتئین برای ساخت یک معماری خاص برای DNA هدف با اصلاح حوزه اتصال به DNA انگشت روی روی یا TALE است. اصلاح پروتئین در ۳ تا ۹ انگشت برای یک محل هدف، یا مهندسی تکرارهای متعدد TALE برای طراحی یک حوزه اتصال به DNA با هدفمندی خاص، چالشبرانگیز، بسیار وقتگیر و پرهزینه است.
CRISPR/Cas9 به دلیل سادگی طراحی، ویژگی هدفمندی بالا، کاهش سمیت خارج از هدف، قابلیت هدف قرار دادن چندین ژن، هزینه کم و سهولت انتقال به سلولها، نسبت به ZFNها و TALENها مزایای متعددی دارد. با این حال، استفاده از CRISPR/Cas برای اهداف درمانی به دلیل اثرات خارج از هدفی که همچنان وجود دارد، نیازمند تحقیقات بیشتر برای تأیید ایمنی آن است. اثرات خارج از هدف با فرکانس بالا، مانعی چالشبرانگیز است که برای دستیابی به ویژگی بالاتر و دقیقتر این روش نسبت به آنچه در حال حاضر امکانپذیر است، باید حل شود. استراتژیهای زیر توسط محققان مختلف برای جلوگیری از اثرات خارج از هدف CRISPR/Cas اتخاذ شده است:
تزریق مستقیم نوکلئازهای Cas9 در سلولهای هدف به جای بیان مجدد پروتئین برای کاهش فعالیت نوکلئاز. استفاده از دو sgRNA برای هدف قرار دادن هر دو رشته DNA توالی هدف همراه با یک نوع نیکزننده DNA از Cas9. این استراتژی ایجاد شکاف دوتایی کاهش قابل توجهی در اثرات خارج از هدف نشان داده است. کوتاه کردن ۲ تا ۳ نوکلئوتید از sgRNA، ویژگی هدفمندی بالای CRISPR/Cas را نشان داده است. چو و همکارانش با افزودن دو نوکلئوتید گوانین در انتهای ۵’ کنار ناحیه متمم هدفمند sgRNA، کاهش اثرات خارج از هدف را نشان دادهاند.
علاوه بر اثرات خارج از هدف، مانع دیگر برای CRISPR/Cas، انتقال کلی این سیستم به سلولهای یوکاریوتی است. چندین سیستم بردار ویروسی و غیر ویروسی برای انتقال Cas9 و sgRNA به صورت in vivo (درون بدن زنده) امیدوارکننده به نظر میرسند. یک استراتژی ex vivo (خارج از بدن زنده) که در آن سلولهای بیمار با CRISPR/Cas به صورت in vitro (در محیط کشت) اصلاح میشوند و سپس دوباره به بیمار پیوند زده میشوند، از نظر انتخاب سلولهای اصلاحشده به درستی و پیوند مجدد بعدی بدون جهشهای خارج از هدف ناخواسته، مزیت قابل توجهی را به همراه دارد. از این روش با موفقیت برای اصلاح سلولهای B به منظور بهبود ایمنی و برای سلولهای T در درمان سرطان استفاده شدهاست. تلاشها برای استفاده از CRISPR/Cas برای اهداف تشخیصی بسیار موفق بوده است. با تلاشهای مداوم برای بهبود سیستمهای شناخته شده CRISPR/Cas و کشف انواع جدیدی از سیستمهای CRISPR، به نظر میرسد که این فناوری در آیندهای بسیار نزدیک برای کاربردهایی از زیستفناوری تا درمان، ایمن و مؤثر خواهد بود.