



فلوسایتومتری خون یک روش آزمایشگاهی است که برای شناسایی، شمارش و ویژگییابی سلولهای خونی استفاده میشود. این روش بر اساس اندازهگیری ویژگیهای فیزیکی و شیمیایی سلولهای خونی با استفاده از پراکندگی نور و فلورسانس استوار است. تشخیص بیماریهای خونی فلوسایتومتری…
فلوسایتومتری یک روش آزمایشگاهی است که برای شناسایی، شمارش و ویژگییابی سلولها یا اجزای سلولی استفاده میشود. این روش بر اساس اندازهگیری ویژگیهای فیزیکی و شیمیایی سلولها با استفاده از پراکندگی نور و فلورسانس استوار است. فلوسایتومتری یک روش قدرتمند…
فلوسایتومتری یک روش آزمایشگاهی است که برای شناسایی، شمارش و ویژگییابی سلولها یا اجزای سلولی استفاده میشود. این روش بر اساس اندازهگیری ویژگیهای فیزیکی و شیمیایی سلولها با استفاده از پراکندگی نور و فلورسانس استوار است. فلوسایتومتری یک روش قدرتمند…
فایل فلوسایتومتری pdf 1 زبان : فارسی تعداد صفحات : 14 حجم فایل فایل فلوسایتومتری pdf 1 زبان : فارسی تعداد صفحات : 31 حجم فایل : 2MB فایل فلوسایتومتری pdf سوم: زبان : فارسی تعداد صفحات : 17 حجم…
فلوسایتومتری یک روش آزمایشگاهی است که برای اندازه گیری ویژگی های فیزیکی و شیمیایی سلول ها یا ذرات استفاده می شود. این روش از یک دستگاه به نام فلوسایتومتر استفاده می کند که جریانی از سلول ها یا ذرات را…
ریل تایم پی سی آر یک تکنیک مولکولی است که برای تعیین کمی میزان DNA یا RNA در نمونه ها استفاده می شود. این تکنیک بر اساس اصل واکنش زنجیره ای پلیمراز (PCR) است که در آن DNA یا RNA…
چکیده و جمع بندی فلوسایتومتری فلوسایتومتری یک تکنیک آزمایشگاهی است که برای اندازه گیری و آنالیز ویژگی های فیزیکی و شیمیایی سلول ها یا ذرات استفاده می شود. این تکنیک بر اساس پراکندگی نور توسط سلول ها یا ذرات و…
ساترن بلات چیست؟ تکنیک آزمایشگاهی Southern Blotting یک روش مولکولی است که برای تشخیص و جداسازی DNA در نمونههای مختلف استفاده میشود. در این روش، DNA نمونه با استفاده از آنزیمهای خاص بریده میشود و سپس با استفاده از الکتروفورز،…
تکنیکهای نوردرن بلات نیز مانند Southern blotting بر پایه هیبریداسیون اسیدهای نوکلئیک هستند و تفاوتشان در آن است که مولکولهای هدف، RNA های هضمنشده (معمولا mRNAها) میباشند. این تکنیکها به منظور سنجش اندازه رونوشتها و نیز دستیابی به الگوی بیان…
نرم افزار اسنپ ژن | SNAPGENE GSL Biotech SnapGene نرم افزار زیست شناسی مولکولی می باشد که استفاده از آن راحت تر از قلم و کاغذ است. اسنپ ژن از نرم افزارهای بسیار خوب برای شبیه سازی مراحل کلون نمودن ژن ها…
نرم افزار AUTODIMER : برنامه های مختلفی برای آنالیز پرایمرها وجود دارد. برنامه AutoDimer یک نرم افزار بیوانفورماتیکی است که می تواند تمام پرایمرهای شما را به طور همزمان آنالیز بکند. این نرم افزار در کاربردهایی که به طور همزمان…

این تکنیک در دهه 1970 توسط دانشمندی به نام Southern Blot برای شناسایی قطعاتی که مکمل یک توالی DNA یا RNA موردنظر بر روی ژل بودند به کار رفت . پس از برش DNA توسط آنزیم محدودالاثر ، قطعات حاصل…

☑️تایید تست تشخیص کرونا ویروس در 45 دقیقه توسط FDA سازمان جهانی دارو و غذا آمریکا، تست Coronavirus rapid که می تواند COVID-19 را در 45 دقیقه تشخیص دهد تایید کرد. این تست که توسط شرکت California-based Cepheid انجام شده،…
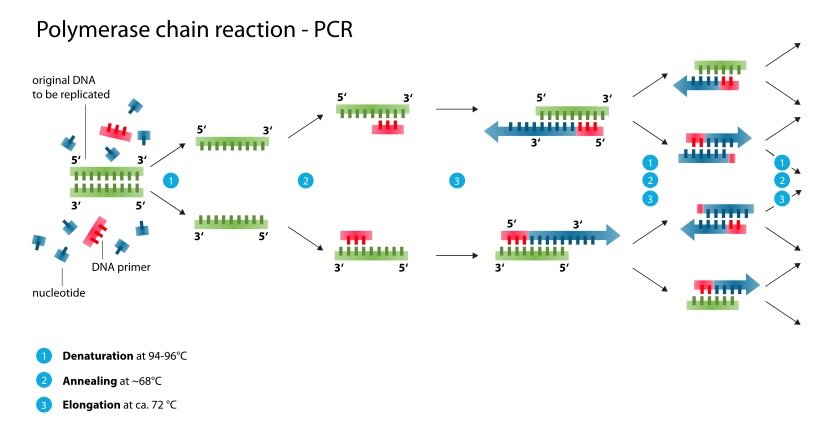
واکنش زنجیرهای پلیمراز (به انگلیسی: Polymerase Chain Reaction) که مخفف آن PCR میباشد. واکنش زنجیرهای پلیمراز (PCR) تکنیکی در زیستشناسی مولکولی است و به منظور تکثیر یک نسخه منفرد یا نسخههای کمی از یک قطعه DNA با توالی خاص به…